Synthèse par Dr Bour,
Dr V.Robert, statisticien, pour l'analyse au chapitre "un amendement apporté à l'étude" - 5 octobre 2023
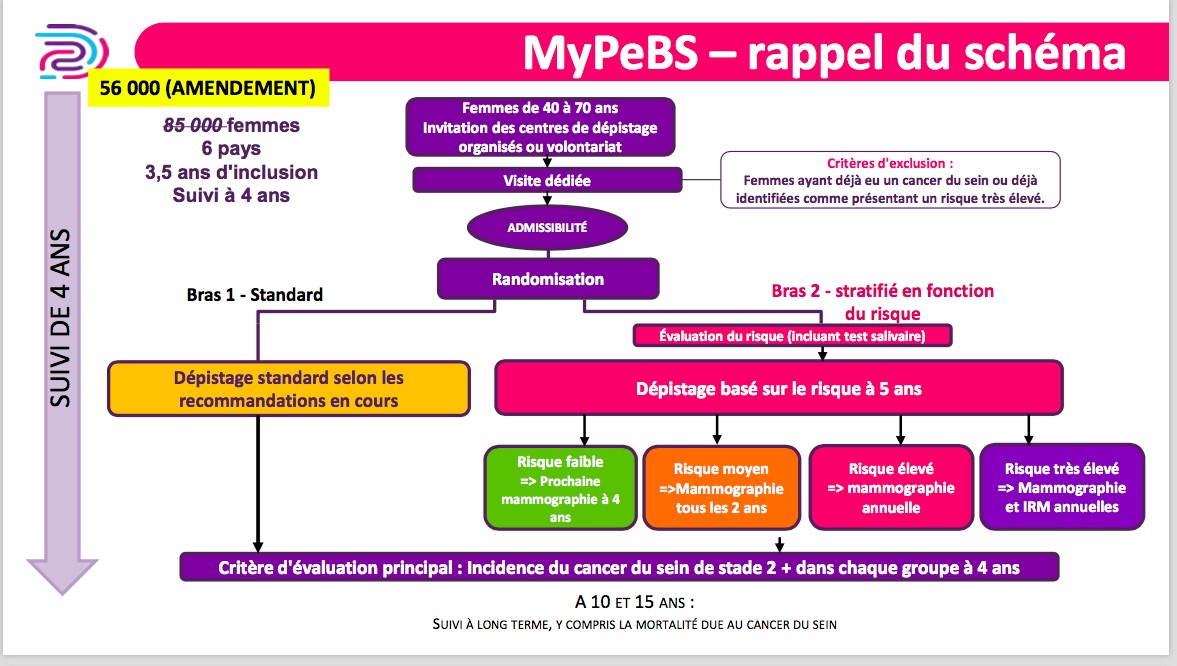
MyPeBS ('My Personal Breast Screening' ou 'mon dépistage personnalisé du cancer du sein') est une étude clinique internationale affichant pour but de comparer une stratégie de dépistage personnalisée au dépistage standard en vigueur.
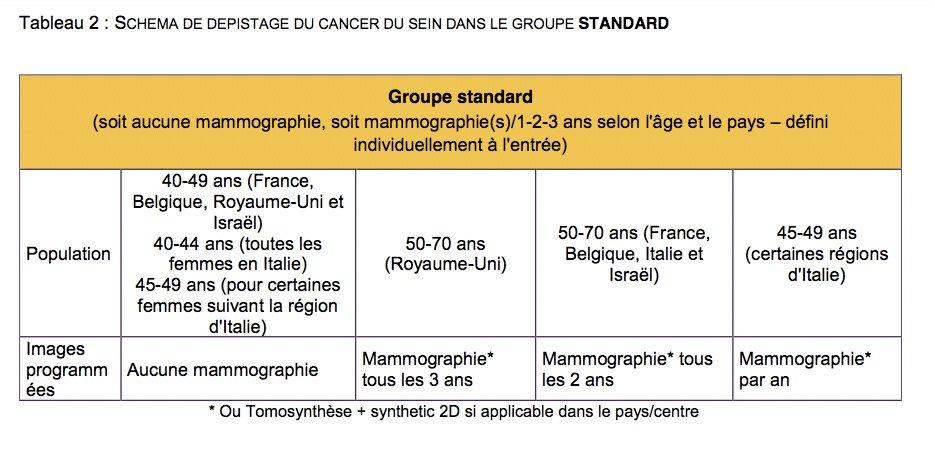
Initialement 85 000 puis 80 000 femmes volontaires âgées de 40 à 70 ans devaient être recrutées dans 5 pays: Belgique, France, Israël, Italie et Royaume-Uni.
Le recrutement a posé beaucoup de problèmes, l'Espagne a été donc rajoutée comme pays recruteur lors de la dernière année, afin de pallier au problème de participation.
Malgré cela, lors de l'arrêt de la phase de recrutement (août 2023), seules 53 142 femmes étaient intégrées dans l'étude.
Site officiel de MyPEBS
Rappel des objectifs de l'étude MyPEBS
Il s'agit d'une étude dont l'objectif principal est de comparer le nombre de nouveaux cas de cancers du sein avancés dans deux groupes de femmes.
Un groupe est composé de femmes soumises à un dépistage adapté à leur niveau de risque personnel de cancer du sein.
L'autre groupe de femmes sera soumis au dépistage organisé habituel.
Non-inférieur ?? Késako....
Il s'agit d'une étude dite de non-infériorité, ce qui signifie qu'il ne s'agit pas du tout de vérifier si le dépistage individuel serait meilleur que le dépistage standard.
Il s'agit de vérifier si le nouveau dispositif (dépistage individualisé) ne serait pas moins bon que le dépistage standard pour détecter des cancers de stade 2 et plus, en tolérant une certaine perte d'efficacité, à hauteur d'un certain seuil tolérable, qu'on appelle le seuil de non-infériorité.
Ici le seuil choisi est de 25%.
Les promoteurs de l’essai expliquent qu’ils s’attendent à trouver 480 nouveaux cas de tumeurs graves pour 100 000 femmes dans le groupe dépistage habituel. Si ce même taux ne dépasse pas 600 pour 100 000 femmes dans le groupe "adapté au risque", les deux groupes seront déclarés équivalents.
Cela signifie que si le taux de cancers graves est augmenté de moins de 25% (par exemple de 18%) dans le groupe "adapté au risque", alors l’étude sera un succès et affirmera que les nouvelles modalités de dépistage sont « aussi efficaces » que les anciennes. Alors qu'un dépistage est censé au départ diminuer drastiquement les formes graves lorsqu'il est vraiment performant...
Les essais de non infériorité sont des études utilisées dans le cas où une intervention donnée est reconnue efficace, mais est associée à des toxicités importantes, et qu'on veut démontrer qu’une nouvelle intervention alternative (celle qui va être testée) est au moins aussi efficace, mais généralement associé à une diminution des toxicités. Et surtout qu'il y a un avantage compensateur pour les patients.
Malheureusement l'étude MyPEBS comporte de nombreux travers que nous avions pointés du doigt.
Et le rationnel de l'étude (protocole qui justifie l'étude,
également) faisait déjà l'aveu suivant :et que nous avons téléchargé
"À ce jour, les dommages supplémentaires (mammographies faussement positives, possibles surdiagnostics, biopsies rétrospectives inutiles, mammographies faussement négatives) et les bénéfices supplémentaires de l’utilisation de l’information sur les risques polygéniques afin d'adapter les stratégies de dépistage (décès par cancer du sein évités, années de vie sauvées ajustées à la qualité de vie, réduction de la mortalité par cancer du sein) demeurent non testées et inconnues." ( page 47, point 1.1.25)
Ce qui n'est pas rassurant.
Un amendement apporté à l'étude
Du fait de l'échec de recrutement de suffisamment de femmes, un amendement (pour l'instant confidentiel) a dû être apporté à la méthodologie et au plan d'analyses statistiques de l'étude, document que nous avons pu consulter.
Par ailleurs nous disposons également d'un document PDF relatif à un webinaire de juin 2022 que nous pouvons reproduire, et il mentionne les informations de ces modifications contenues dans l'amendement.
PDF webinaire juin 2022 : MyPeBSwebinaire300622
Slide 9
Par Dr V.Robert
En raison de difficultés de recrutement, le nombre de femmes à inclure dans l'étude a dû être revu à la baisse.
Comment les promoteurs de l'étude justifient-ils cette baisse des inclusions ?
- En changeant de stratégie d'analyse des résultats.
Initialement, l'analyse était prévue en per-protocole. Autrement dit, seules les participantes ayant parfaitement respecté le protocole de l'étude pouvaient être analysées. Ce mode d'analyse a une conséquence évidente : les femmes n'ayant pas respecté le protocole sont comptées dans les inclusions mais ne comptent pas dans l'analyse. Il faut donc inclure plus de femmes que le nombre nécessaire pour l'analyse. Le protocole initial prévoyait 42 500 inclusions dans chaque bras, avec 30% de "pertes" par non-respect du protocole + perdues de vue dans le bras dépistage personnalisé et 10% de "pertes" dans le bras dépistage standard (anticipated rate of non-compliance). On devait donc avoir 85000 femmes incluses (2x42500) et seulement 68000 femmes analysables (42500 – 42500x30/100 = 29750 dans le groupe dépistage personnalisé et 42500 – 42500x10/100 = 38250 dans le groupe dépistage standard).

Slide 40
L'analyse en per-protocole a été abandonnée au profit d'une analyse en intention de traiter. Dans une analyse en intention de traiter, toutes les participantes sont analysables (même les "non-compliantes"), même si elles n'ont pas respecté le protocole de l'étude. Du coup, il y a moins de "pertes". Les promoteurs de MyPeBS estiment à 5% (dropout rate) , dans chaque bras, les "pertes" par perdues de vue. Concrètement, cela signifie que, pour avoir 68000 femmes analysables, il n'y a plus besoin d'inclure que 71600 femmes (au lieu des 85000 du protocole initial) (explication : 71600 – 71600x5/100 = 68000).
- En augmentant la différence supposée entre les 2 bras.
Comme 71600 femmes à inclure, c'est encore au-dessus des capacités d'inclusion, les promoteurs utilisent une autre astuce pour réduire le nombre d'inclusions nécessaires : ils font passer la différence supposée entre les 2 bras de 10% à 12,5% (relative improvement).
Pour comprendre l'intérêt de cette astuce, il faut se rappeler que chercher une différence entre 2 groupes, c'est un peu comme chercher une aiguille dans une botte de foin. Plus l'aiguille (la différence entre les groupes) est grosse, plus l'aiguille sera facile à trouver, et moins on aura besoin d'une grosse loupe (le nombre de femmes incluses). En postulant une différence de 12,5% plutôt que 10%, les promoteurs parient sur une plus grosse "aiguille" et ont besoin d'une plus petite "loupe". Cette 2ème astuce leur permet de "justifier" 56300 inclusions.
Que penser de ces justifications ?
L'analyse en intention de traiter est habituellement moins puissante mais comporte moins de risque de biais qu'une analyse en per-protocole. On peut donc facilement admettre le passage d'une analyse en per-protocole vers une analyse en intention de traiter. On peut juste s'étonner que le choix d'une analyse en intention de traiter n'ait pas été fait dès le protocole initial.
Mais le plus surprenant est l'augmentation de la différence supposée de 10 à 12,5%. Comme mentionné ci-dessus, une analyse en intention de traiter est habituellement moins puissante qu'une analyse en per-protocole (les violations du protocole conduisent à atténuer l'éventuelle supériorité d'un des bras). La logique aurait donc été de revoir à la baisse la différence supposée entre les 2 bras et certainement pas de l'augmenter.
L'explication de ces choix illogiques est évidente. Normalement, une méthodologie correcte impose de fixer le type d'analyse et l'efficacité supposée et ensuite, et seulement ensuite, de calculer les effectifs à inclure. Ici, les promoteurs de MyPeBS font exactement le contraire : ils fixent les effectifs à inclure en fonction de leur capacité d'inclusion puis ils bidouillent le type d'analyse et l'efficacité supposée jusqu'à ce que ça colle avec les effectifs prédéfinis.
On est bien loin de la rigueur qui devrait être respectée lors d'un essai clinique et cette magouille méthodologique discrédite complètement les futures conclusions de l'étude, quelles que soient ces conclusions. Les promoteurs ne sont pas responsables de l'épidémie COVID mais l'honnêteté scientifique aurait été, soit de prolonger les inclusions jusqu'aux 85000 initialement prévues, soit de jeter l'éponge et d'arrêter l'étude faute de capacité à inclure un nombre suffisant de participantes.
Problème d'une trop grande homogénéité des pannels
Le recrutement n'est pas suffisamment varié pour représenter la population, comme les promoteurs eux-mêmes s'en émeuvent dans un mail du mois de mai 2023 envoyé aux investigateurs..

L'interprétabilité des résultats est mise à mal si dans la population des femmes recrutées une catégorie socio-professionnelle se retrouve sur-représentée, ces résultats ne correspondent qu'à une catégorie de femmes d'un niveau économique particulier, et les conclusions de l'étude seront difficilement généralisables sur une population réelle bien plus diverse.
En conclusion
De toute évidence, la révision des effectifs à inclure n'est pas motivée par des considérations statistiques mais par la difficulté à inclure les femmes dans l'étude. Plutôt que de "bidouiller" le protocole pour tenter de justifier la diminution des effectifs, il eut été plus honnête de reconnaître l'échec et d'abandonner le projet.
Que l'ANSM, un CPP et UNICANCER aient accepté le "bidouillage" ne contribue pas à la crédibilité de ces institutions.
Enfin, ne l'oublions pas, cette étude ne disposant pas d'un groupe sans dépistage, elle permet de donner un seul choix aux femmes, entre un dépistage et un autre ; elle permet seulement de conclure que quelle que soit la forme de dépistage, l'un n'est pas moins bon que l'autre.
Cela permet ainsi de faire disparaitre la notion d'absence de dépistage du discours, et d'une hypothèse envisageable d'un non-dépistage tout aussi acceptable.