Un logiciel dédié à MyPebs, appelé Mammorisk a été conçu spécialement pour l'étude MyPebs
document MammoRisk_Briefpresentation_2018
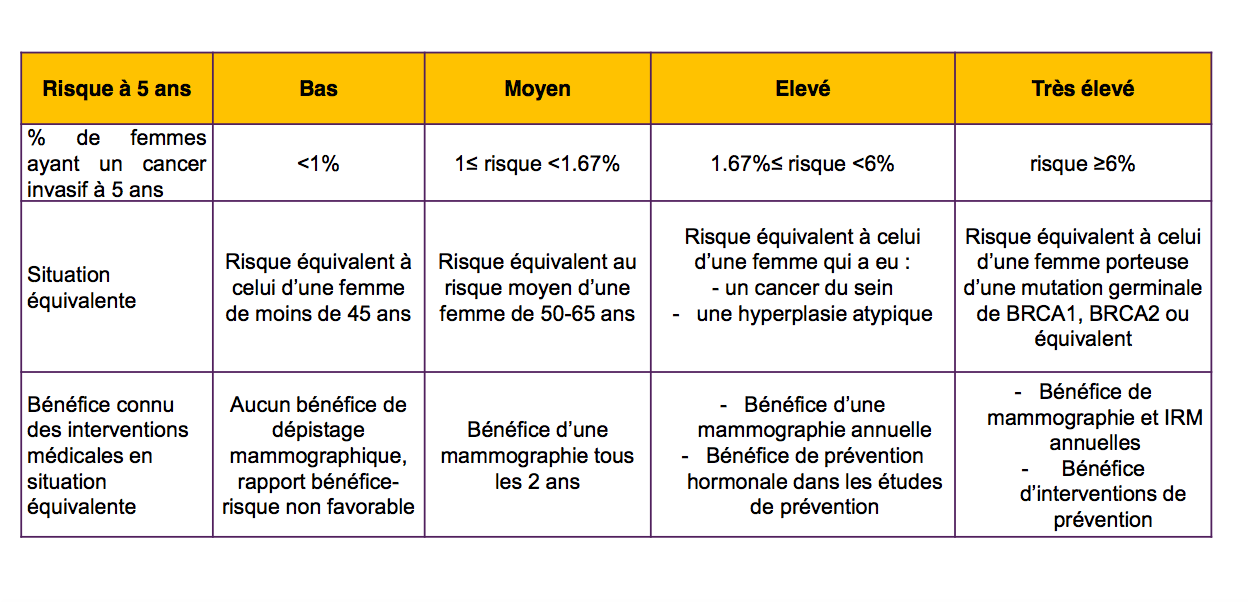
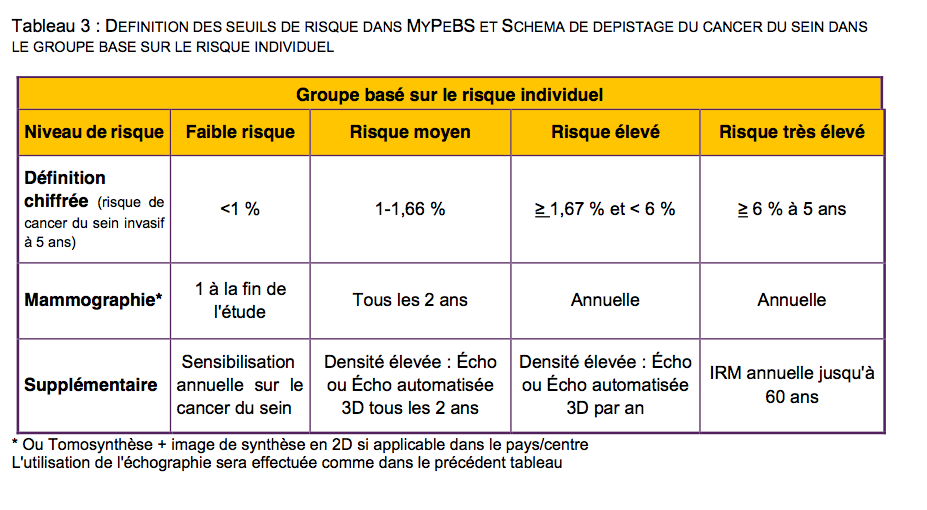
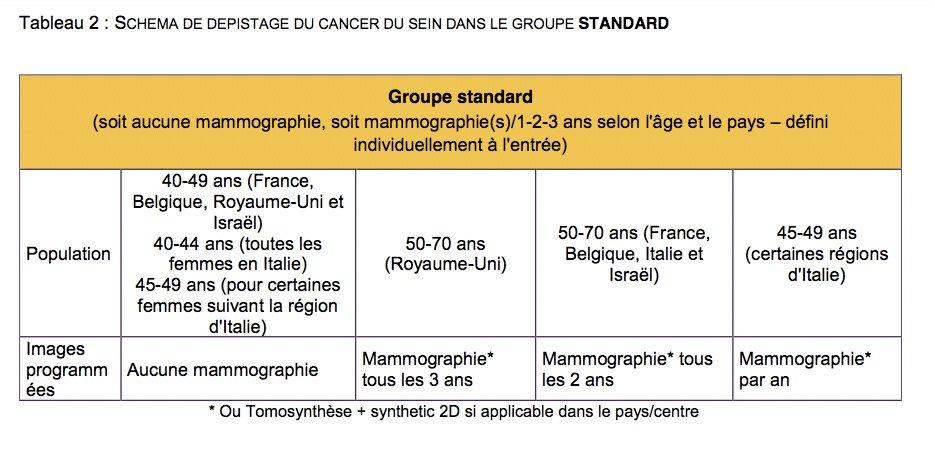
Dans l'étude MyPEBS, pour "stratifier" le risque, celui-ci sera évalué selon un algorithme, défini par le comité directeur de l'étude (c'est à dire la coordinatrice de l'étude : Dr Suzette Delaloge, chef du comité de pathologie mammaire de Gustave Roussy ; et l'investigateur principal en France : Dr Corinne Balleyguier, chef du service d'imagerie diagnostique de Gustave Roussy ).
Page 12 du synopsis de l'étude MyPebs (MyPEBS SYNOPSIS . pdf ) :

Ce logiciel Mammorisk inclut donc comme facteurs de risque :
- l'âge,
- les antécédents familiaux,
- les antécédents de biopsie bénigne antérieure,
- la densité à la mammographie,
- ainsi que les résultats génétiques.
Voici le schéma du logiciel :
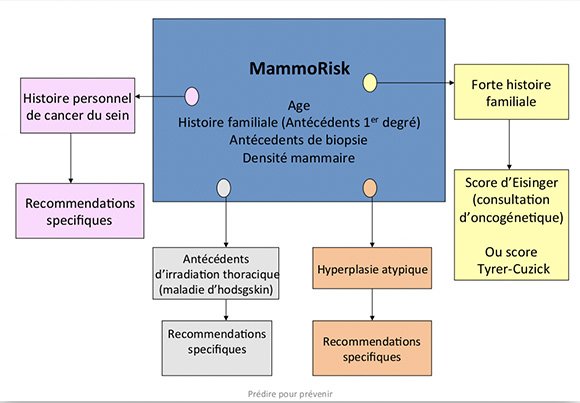
1. Analyse des critères choisis
A- la densité mammaire
Plusieurs études sont disponibles, depuis l'étude de Wolfe[1] sur la relation entre densité mammaire et risque de cancer du sein. Cette étude ancienne a été fortement contestée à l'époque, même par les tenants du dépistage.
D'autres études ont vu le jour depuis, étudiant la pertinence de relier ce facteur de densité avec d'autres facteurs de risque, pour pouvoir élaborer des modèles de calculs du risque de contracter dans les 5 ans un cancer du sein.[2] [3] [4] [5] [6]
Aujourd’hui, aucun outil d’estimation du risque de cancer du sein utilisant la densité mammaire n’a, pour l’heure, fait la preuve de sa pertinence.
Une annonce sur le site de la société Predilife qui a conçu le logiciel attire notre attention [7]: " MammoRisk mesure la densité mammaire qui est un facteur de risque important pour la femme de plus de 40 ans. "
Qu’en est-il d’une telle affirmation ?
La HAS, dans un travail sur l’identification des facteurs de risques, écrit :
« La densité mammaire élevée avant la ménopause n’a pas été retenue comme un facteur de risque à l’issue des travaux du volet 1 ».
La société va donc clairement à l’encontre de l’HAS qui ne reconnaît pas la densité mammaire comme facteur de risque.
De plus, pour utiliser cet outil d’estimation du risque, il faut pratiquer une mammographie si aucune n’a été réalisée, et cela dès 40 ans.
Là aussi, cela va à l’encontre des recommandations de l’HAS qui ne reconnaît la mammographie comme utile qu’à partir de 50 ans. [8].
Enfin, la réalisation d’un examen radiologique irradiant, au seul prétexte de se conformer à un outil informatique, est-elle éthique ?
C’est une question que personne, ni parmi les investigateurs ni parmi les coordonnateurs de cette étude ne semble pas s’être posée, pas plus d’ailleurs que le autorités sanitaires qui ont autorisé cette étude.
B- l'âge
L'âge est reconnu depuis longtemps comme facteur de risque du cancer du sein avec un pic statistique entre 50 et 60 ans.
C- les antécédents
Les deux autres facteurs inclus dans le logiciel, antécédents de biopsie du sein même bénigne, et antécédents familiaux de cancer mammaire prêtent aussi à discussion.
Avec le surdiagnostic que les évaluations les plus récentes et fiables situent aux alentours de 40%,( jusqu'à même 50%), le dépistage a généré de la maladie, et a ainsi augmenté artificiellement le nombre des familles à risque.
Le nombre d'actes biopsiques chez des femmes jeunes pour des lésions bénignes telles que des fibro-adénomes, en vue d'une exérèse chirurgicale, a très nettement augmenté ces dernières années.
Il le sera d’autant plus, dans l’avenir, que la consultation systématique proposée aux femmes dès 25ans, vient d’être instaurée [9].
A 25 ans, les lésions cancéreuses sont rarissimes, mais les lésions bénignes (kystes, mastose, fibro-adénomes) sont fréquentes et facilement biopsiées sous échographie.
Tous ces éléments montrent une augmentation des examens des seins chez les femmes jeunes.
On peut donc sans risque s’inquiéter sur le fait que de très nombreuses femmes se verront attribuer un facteur de risque de cancer du sein au seul prétexte d’examens et de biopsies plus liés au « toujours plus médical » qu’à de réelles problématiques de santé.
2. La validation scientifique du logiciel Mammorisk
Sur le site de la société Predi-Life (ou Statlife) n'apparaît aucune source bibliographique.
En revanche à la page 13 du document de présentation du logiciel il y a sous le titre de "scientific papers" trois sources bibliographiques :

Seule la première référence a été publiée :
Laureen Dartois et al, A comparison between different prediction models for invasive breast cancer occurrence in the French E3N cohort, Breast Cancer Research and treatment, 2015. Les deux autres, pas encore.
A cette étude contribuent Mme Suzette Delaloge, coordonnatrice de l'étude MyPEBS ainsi que Mr Emilien Gauthier, qui est le directeur de recherche et de développement pour Mammorisk de la société Predilife [10] .
La deuxième référence n’a pas été publiée dans une revue médicale. Le texte indique que l’étude est « in press » en 2017 dans le European Journal of Cancer. En réalité, comme le montre une recherche faite sur le site de ce journal au 16 avril 2019, aucune étude signée par Mr Ragusa n’a été publiée dans cette revue en 2017, ni 2018, ni 2019. Le plus probable est que la publication a été refusée à la suite de la revue par les pairs.
Son contenu correspond cependant à un poster présenté au Symposium de San Antonio de 2016, dont les auteurs principaux sont Mr Stéphane Ragusa, président et créateur de la société Predi-Life et Mr Emilien Gauthier, sus-cité.
Comme co-auteure nous retrouvons Mme Suzette Delaloge, oncologue de l’Institut Gustave Roussy et promoteure de l’étude MyPEBS. Il est important de noter que les communications dans les symposiums ne font pas l’objet d’une revue par les pairs et n’ont pas la même valeur qu’une publication dans une revue médicale.
(Sujet du poster
« Développement et validation d’un nouveau modèle non paramétrique d’évaluation des risques de cancer du sein sur les populations américaines et européennes de dépistage. »)
La troisième référence : l'étude RIVIERA
« RIVIERA - Evaluation du niveau de risque de cancer du sein chez des femmes de la population générale par leur médecin de ville: faisabilité, ressenti, acceptabilité, satisfaction, adhésion aux programmes de suivi. »[11] [12] .
L'investigatrice principale est Mme Delaloge.
L'essai inclut 600 femmes et est effectuée en collaboration avec la société Statlife, et avec le partenariat de l'Institut Gustave Roussy.
Elle est censée analyser "l'acceptabilité et la faisabilité d'une consultation de prévention du cancer du sein par les médecins de "cabinets de ville" - radiologues, gynécologues, généralistes - en utilisant MammoRisk, une solution logicielle innovante de prédiction et de prévention du risque de cancer du sein".
Riviera est promue par l'IGR (Institut Gustave Roussy) comme cela est précisé dans le descriptif de l'étude (voir référence 11) et financée par l'ARC (Fondation pour la Recherche contre le Cancer).[13]
« Il s'agit d'une étude nommée de "soins courants", qui permettra, si l'étude est positive, de proposer une possible généralisation de ce logiciel chez les médecins de ville pour une "prévention personnalisée" du cancer du sein et une extension des ventes du logiciel ».
Mais en quoi l’acceptation par les femmes et la faisabilité d’une consultation de prévention qui utilise le Mammorisk donne des indications sur l’intérêt de ce logiciel ?
En d’autres terme, ce n’est pas parce que vous êtes d’accord avec quelque chose qui vous paraît "acceptable", que ce quelque chose est « valable » et valide son intérêt.
Au total il n’apparaît rien, sur les études scientifiques présentées par les promoteurs et concepteurs du Mammorisk, qui en valide l’intérêt.
Il n’y a de référence que celles des concepteurs de l’étude et quasiment aucune d’auteurs indépendants de ce Mammorisk.
Nouvelle étude publiée en 2022
Là aussi une étude de faisabilité.
Elle très peu de participantes (290), et qui consultent en raison d'un sur-risque supposé ce qui constitue déjà un biais.
Elle est réalisée par Saghatchian et 'coll'. Mme Saghatchian a reçu des honoraires de la société Predilife commercialisant Mammorisk. comme indiqué dans la déclaration d'intérêts.

Examinons quelques-uns des collaborateurs :
Mr Emilien Gauthier, co-auteur, n'est autre que le directeur de recherche et de développement pour Mammorisk de la société Predilife.
Une recherche nous apprend que Mme Valérie Hélin, également parmi les co-auteurs, est superviseur des affaires médicales et réglementaires chez Statlife, (marque semi-figurative de Predilife, Statlife est définie comme une société de medtech qui développe des logiciels médicaux de prédiction de risque).
Deux études de faisabilité donc, toutes les deux supportées par la société commercialisant le logiciel, pour "valider" Mammorisk....
3.un schéma pour mieux comprendre les intrications
MMe Suzette Delaloge est présidente du groupe French Breast Cancer Intergroup - UNICANCER (UCBG)[14] qui est partenaire de l'étude MyPEBS dont Mme Delaloge est la coordinatrice principale ; elle est oncologue, chef du comité de pathologie mammaire à l'IGR, investigatrice principale de l'étude RIVIERA, faite en partenariat avec la société Statlife qui commercialise le logiciel Mammorisk intégré dans l'étude MyPEBS pour laquelle MMe Delaloge est la coordinatrice principale.
La société Statlife (ou Predilife) et l'IGR sont partenaires de l'étude Riviera qui valide l'acceptabilité du Mammorisk produit par la société Statlife (ou Predilife), cette étude ainsi que le logiciel sont financés par l'association ARC.[15]
L'IGR est membre d'Unicancer.[16]
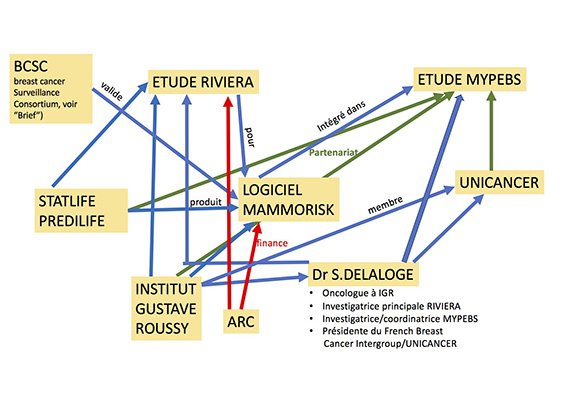
Nous voyons assez facilement grâce au schéma comment des coordonnateurs d'études et des institut peuvent promouvoir des études permettant l’intégration de dispositifs médicaux avec une validation scientifique a minima et auto-promue, émanant d'une start-up privée, afin d'intégrer leur produit dans une étude européenne à grande échelle financée par les deniers de l'Union Européenne.
Ci-dessous d'autres renseignements utiles sur « la success story à venir » de la société Predilife, ainsi qu'une capture d'écran trouvée dans la revue "Le Revenu".

4. Position de la Haute Autorité de Santé
"Seuls les modèles intégrant les facteurs de risque individuels sont exploitables pour déterminer des populations à risque accru (ou diminué) de cancer du sein. La comparaison avec le risque en population générale permet de cibler des populations pouvant faire l’objet de recommandations spécifiques. Toutefois, la capacité des modèles à prédire la survenue de cancer du sein reste médiocre (indice de concordance autour de 0,65). Tous les facteurs de risque ne sont pas pris en compte, notamment les antécédents médicaux personnels, la contraception hormonale, la consommation d’alcool ne sont pas inclus dans les modèles. Enfin, les outils ne sont pas disponibles pour toutes les populations (femmes de moins de 35 ans, femmes américaines hispaniques, etc.). A notre connaissance, ces modèles n’ont pas fait l’objet de validation dans la population française, et l’article de de Pauw et al. (31) montre que, pour une même femme présentant trois antécédents familiaux de cancers du sein, les différents modèles estiment des risques de survenue de cancer du sein très différents, de 13 à 34 %. Ces modèles ne sont pas fournis avec des grilles de lecture et des algorithmes de décision pour le clinicien, permettant de choisir une stratégie de surveillance en fonction de l’estimation obtenue."
Page 53
5.Conclusion
Dans toute cette analyse, une question essentielle se pose : où est l’intérêt des femmes, qui éblouies, sans doute, par les termes « innovation » « médecine personnalisée » etc ; « confieront» leurs seins à l’étude MyPeBS ?
Après avoir analysé en détail et critiqué le formulaire de consentement présenté aux femmes, le protocole de cette étude et la problématique de la non-infériorité, nous nous posons aujourd’hui la question du réel l’intérêt scientifique de l’étude MyPeBS par l’analyse du logiciel Mammorisk qui est la « pierre angulaire » de cette étude.
Cette étude n’a-t-elle pas un but marketing et de développement financier plutôt que scientifique comme présenté : « MyPeBS : mobilisation européenne pour proposer un dépistage personnalisé, plus efficace et plus sûr » [17] ?
BIBLIO
[1] Wolfe JN. Breast patterns as an index of risk for developing breast cancer. AJR 1976;126:1130-9.
[2] Annals of Internal Medicine Personalizing Mammography by Breast Density and Other Risk Factors
for Breast Cancer: Analysis of Health Benefits and Cost-Effectiveness
John T. Schousboe, MD, PhD; Karla Kerlikowske, MD, MS; Andrew Loh, BA; and Steven R. Cummings, MD
[3] https://www.researchgate.net/publication/273154592_The_Contributions_of_Breast_Density_and_Common_Genetic_Variation_to_Breast_Cancer_Risk
The Contributions of Breast Density and Common Genetic Variation to Breast Cancer Risk
Article (PDF Available) in JNCI Journal of the National Cancer Institute 107(5) · May 2015 with 77 Reads
DOI: 10.1093/jnci/dju397 · Source: PubMed
[4] McCormack VA, dos Santos Silva I. Breast density and parenchymal patterns as markers of breast cancer risk: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;15(6):1159–1169
[5] KERlikowske K, Cook AJ, Buist DS, et al. Breast cancer risk by breast density, menopause, and postmenopausal hormone therapy use. J Clin Oncol. 2010;28(24):3830–3837.
[6] https://link.springer.com/article/10.1007/s10549-011-1853-z
Breast Cancer Research and Treatment
May 2012, Volume 133, Issue 1, pp 1–10| Cite as
Risk prediction models of breast cancer: a systematic review of model performances Thunyarat Anothaisintawee, Yot Teerawattananon, Chollathip Wiratkapun
[7] https://mammorisk.com/fr/documentation-mammorisk/#
[8] https://www.has-sante.fr/portail/jcms/c_1741170/fr/depistage-du-cancer-du-sein-en-france-identification-des-femmes-a-haut-risque-et-modalites-de-depistage
La HAS rappelle qu’en l’absence des facteurs de risque pour lesquels un dépistage spécifique du cancer du sein est recommandé, il n’y a pas lieu de réaliser une mammographie ou une échogra- phie mammaire de dépistage en dehors de la tranche d’âge de participation au programme national de dépistage organisé, c’est-à-dire entre 50 et 74 ans.
[9] https://solidarites-sante.gouv.fr/archives/archives-presse/archives-communiques-de-presse/article/marisol-touraine-modernise-le-depistage-organise-du-cancer-du-sein-et-annonce
[10] https://mammorisk.com/fr/societe-predilife/#
[11] https://www.gustaveroussy.fr/fr/riviera-resultats-positifs-mammoriskr-depistage-cancer-sein
[12] https://mammorisk.com/fr/etude-riviera-mammorisk/#
[13] https://www.fondation-arc.org/actualites/gustave-roussy-presente-resultats-positifs-etude-clinique-riviera-mammorisk
[14] http://www.unicancer.fr/la-recherche-unicancer/french-breast-cancer-intergroup-unicancer-ucbg
[15] https://mammorisk.com/fr/societe-predilife/
[16] https://www.gustaveroussy.fr/fr/gouvernance-generalites
[17] http://www.unicancer.fr/sites/default/files/MyPeBS-DP.pdf